(Grab a refreshment, this is going to be a long one)
Looking more closely at your usr_rates_des.f we found a few more problems. Here’s the key section, with lines numbered for reference (this is actually at line 129 in your file)
1 IF ( time > 1000) THEN
2 IF(((DES_X_s(NP,Fe3O4_S_SolidA)*MW_S(NP,Fe2O3_S_SolidA))/MW_S(NP,Fe3O4_S_SolidA)) < 1.0d0) THEN
3 DES_RATES(Reaction_1) = 0.01*(2.5d-3 * exp(-3.2475d3/DES_T_s(NP)) * (1.0d0-((DES_X_s(NP,Fe3O4_S_SolidA)*MW_S(NP,Fe2O3_S_SolidA))/MW_S(NP,Fe3O4_S_SolidA)))**(-2.0d0/3.0d0))
4 DES_X_s(NP,Fe3O4_S_SolidA)= DES_X_s(NP,Fe3O4_S_SolidA)+2*DES_RATES(Reaction_1)
5 ENDIF
6 ENDIF
-
Previously mentioned - it is not the job of this function to update mass fractions (DES_X_S) so the line (4) should be removed.
-
MW_S(NP,Fe2O3_S_SolidA) is an improper array access. The array MW_S contains the molecular weight for each species, and the array indices are (phase, species) You can see this in the help text for the keyword:
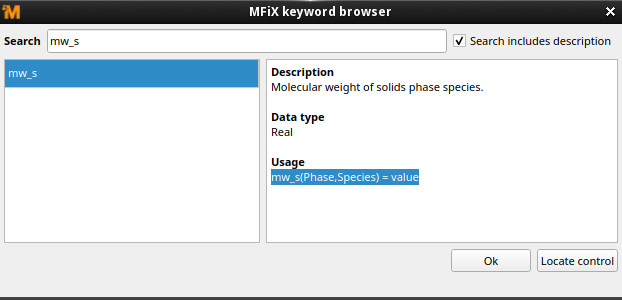
or the keyword reference section of the MFiX user manual
In SUBROUTINE USR_RATES_DES(NP, pM, IJK, DES_RATES),
NP is a particle index, and pM is a phase index. This function is called for each particle in each phase. In your case, there is only 1 phase, and 500 particles. So NP will range from 1 to 500. MW_S(NP,Fe2O3_S_SolidA) will grab some random number out of the computer’s memory, whenever NP is greater than one (99.8% of the time). In a better world, the compiler or Fortran runtime would have caught this out-of-bounds array access… but alas, it does not (perhaps a future MFiX version will have an enhanced DEBUG build which catches stuff like this, but for now it doesn’t).
So
MW_S(NP,Fe2O3_S_SolidA)
must be changed to
MW_S(pM,FE203_S_SolidA)
But this implicitly assumes that pM is always 1 (since you only have one phase defined). That’s OK but if you add a second phase, this code will break. So perhaps there should be a line
if (pM != 1) return
before anything else … this will future-proof you against adding additional phases. If you are sure will never have a second solids phase, this is optional.
Note that species.inc (can be found in the build subdir) contains aliases for the species, but not the named phases:
INTEGER, PARAMETER :: H2_REF_ELEMENT = 1
INTEGER, PARAMETER :: H2O = 2
INTEGER, PARAMETER :: Fe2O3_S_SolidA = 1
INTEGER, PARAMETER :: Fe3O4_S_SolidA = 2
INTEGER, PARAMETER :: Reaction_1 = 1
since you have named your single solid phase Solid you might expect an alias for that, but for various reasons these are not generated (for one thing, there might be name collisions). So you have to use 1 for the phase index, rather than the name Solid.
So instead of writing
MW_S(pM,FE203_S_SolidA)
I would probably add a check for pM=1 and after that just write
MW_S(1,FE203_S_SolidA)
But note that the expression:
MW_S(NP,Fe2O3_S_SolidA))/MW_S(NP,Fe3O4_S_SolidA))
appears several times in your code. So I would suggest:
DOUBLE PRECISION :: MW_RATIO
MW_RATIO = MW_S(1,Fe2O3_S_SolidA)/MW_S(1,Fe3O4_S_SolidA)
- Note the assignment
! Alias particle temperature.
Tp = DES_T_s(NP)
This means that inside the expression, you can write Tp instead of DES_T_s(NP) which helps make the code more readable.
We can also do this for DES_X_s:
! Alias mass fraction
Xs = DES_X_S(NP,Fe3O4_S_SolidA)
-
Break up excessively long lines.
-
Combine multiplicative constants and remove spurious parenthesis
This brings us from
IF( ((DES_X_s(NP,Fe3O4_S_SolidA)*MW_S(NP,Fe2O3_S_SolidA))/MW_S(NP,Fe3O4_S_SolidA)) < 1.0d0) THEN
DES_RATES(Reaction_1) = 0.01*(2.5d-3 * exp(-3.2475d3/DES_T_s(NP)) * (1.0d0-((DES_X_s(NP,Fe3O4_S_SolidA)*MW_S(NP,Fe2O3_S_SolidA))/MW_S(NP,Fe3O4_S_SolidA)))**(-2.0d0/3.0d0))
ENDIF
to
IF (Xs*MW_RATIO < 1.0d0) THEN
DES_RATES(Reaction_1) = 2.5d-5 * &
exp(-3.2475d3/Tp) * &
(1.0d0-(Xs*MW_RATIO))**(-2.0d0/3.0d0)
ENDIF
which is quite a bit easier for me to read and understand.
-
But what if the IF condition is not met? Should we set DES_RATES to 0 in that case?
-
There’s a fair bit of commented-out “junk” code in your usr_rates_des.f and much of it is potentially misleading (e.g. comments about CGS units). Strip out all that stuff about Psat_H2O, CaCO3, etc. There are a large number of USE statements you don’t need. Don’t be afraid to delete code. And indent consistently. It’s just a lot easier to look at the code when it’s tidy. Here’s a somewhat cleaned-up copy of the file:
usr_rates_des.f (972 Bytes)
- After doing the above, there is still a slowdown at 10s when the reactions switch on. But it is not as severe. I suspect that the reaction rates are still too high:
Hope this helps,
– Charles









